尿素‐水系の混ざり方の詳細:
微分的溶液熱力学の応用
One of us has been developing a differential approach in solution
thermodynamics and applying it to studies of aqueous solutions. As a result,
much deeper insights have been gained in molecular processes in aqueous
solutions than hitherto possible. Details are given in a monograph (Y. Koga,
Solution Thermodynamics and Its Application to Aqueous
Solutions: A Differential Approach, Elsevier, Amsterdam (2007)).
Briefly, the mixing scheme was found crucially dependent on the composition.
In the H2O-rich range, the integrity of
H2O is retained in that the hydrogen bond network is
bond percolated and the hydrogen bond energy and angle fluctuate widely. While
solute molecules modify the above characteristics somewhat, the percolation
nature remains intact up to the threshold composition. The detail of
modification depends on the nature of solute. In the solute-rich range, solute
molecules cluster together with a similar local molecular arrangement as in
their pure state with H2O molecules interacting with
such clusters as a single molecule. In the intermediate region, two kinds of
clusters, one rich in H2O and the other in solute
molecules. We call them Mixing Scheme I, II and III from the
H2O-rich end. Such detailed information is gained
directly from the behavior of the second and third derivative quantities that
are obtained without resorting to any fitting function.
Urea - H2O system has been studied extensively due to
its common occurance in biologically important solutions. In 1968, H. S. Frank
and F. Franks advocated that urea is a typical “structure breaker”
as opposed to “structure maker” that forms the so-called
“icebergs” in H2O. Since then, urea's
specific functions towards biopolymers in aqueous solutions have been
attributed to its “structure breaker” properties. Recently,
however, modern spectroscopic studies have caused a great deal of controversy
as to this structure maker/breaker classification. On the other hand, we have
suggested by using our 1-propanol (1P) probing methodology (See Chapters VI and
VII in the monograph cited above) that urea is a typical hydrophile which forms
hydrogen bonds directly to the existing hydrogen bond network of
H2O and retains the network connectivity. At the same
time, however, the large degree of fluctuation inherent in liquid
H2O is reduced progressively by breaking the H
donor/acceptor symmetry enjoyed in pure H2O.
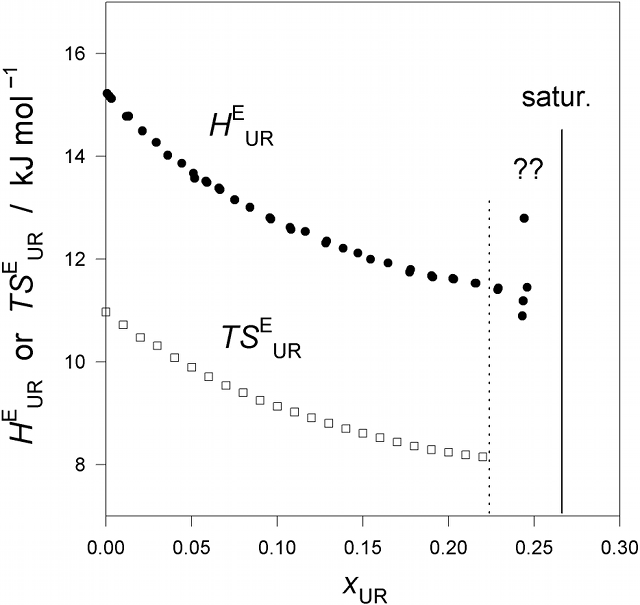
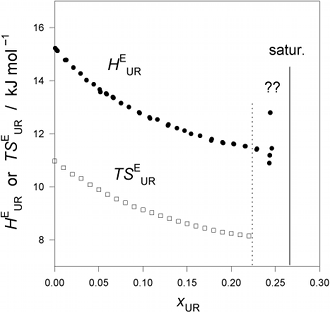 Fig. 1. (Click to enlarge.)
The excess partial molar enthalpy of urea (UR),
HEUR,
and entropy (times T),
TSEUR, in UR - H2O at 25 °C.
The vertical line indicates the saturation point. The region marked by ??
above the vertical dotted line is where dissolution slows down dramatically.
Fig. 1. (Click to enlarge.)
The excess partial molar enthalpy of urea (UR),
HEUR,
and entropy (times T),
TSEUR, in UR - H2O at 25 °C.
The vertical line indicates the saturation point. The region marked by ??
above the vertical dotted line is where dissolution slows down dramatically.
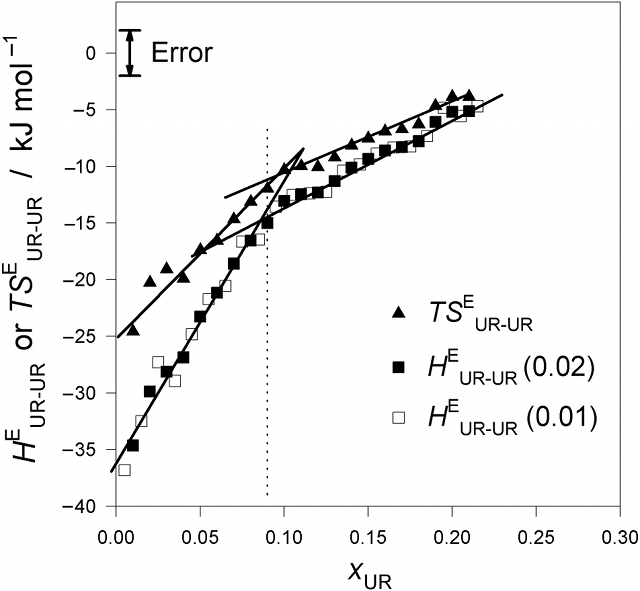
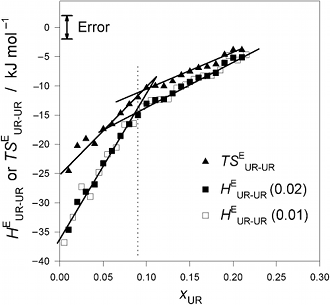 Fig. 2. (Click to enlarge.)
Urea - urea enthalpic and entropic interaction functions,
HEUR-UR and
TSEUR-UR, in UR - H2O at 25 °C.
The break in slope at xUR
≈ 0.09 is evident. The slope changes to about 1/2.7. This value is one of
circumstantial evidences for suggesting dimer or trimer formation. See text.
Fig. 2. (Click to enlarge.)
Urea - urea enthalpic and entropic interaction functions,
HEUR-UR and
TSEUR-UR, in UR - H2O at 25 °C.
The break in slope at xUR
≈ 0.09 is evident. The slope changes to about 1/2.7. This value is one of
circumstantial evidences for suggesting dimer or trimer formation. See text.
Here, we revisit the urea (UR) - H2O system, by
directly determining the excess partial molar enthalpies of UR,
HEUR ( ≡
(∂HE/∂nUR)), a second derivative quantity,
for the first time by using LKB Bromma 8700 isothermal titration calorimeter of
this Center with the ampoule breaking mode. As described below, from the
results together with those from our earlier 1P-probing work, we learned that
urea starts to aggregate at mole fraction of UR
xUR ≈ 0.09, possibly as
dimer or trimer, and yet retain the integrity of H2O
up to xUR ≈ 0.22,
i. e.
the solution is still in Mixing Scheme I. Beyond this composition,
the solution appears to be in Mixing Scheme II and preparing for precipitation
of urea at the saturation, xUR
= 0.266. In the latter region close to the saturation, we observed dramatic
slowing down of UR dissolution. Fig. 1 below shows the results of the excess
partial molar enthalpy of urea,
HEUR, in aqueous urea solution at 25 °C. From another
earlier work (E. C. H. To et al.,
J. Phys. Chem. B 102,
10958 (1998)) in which we determined the excess chemical potential of UR,
μEUR, we calculated the excess partial molar entropy of UR,
SEUR. The values of
TSEUR are also plotted in Fig. 1. We then draw a smooth curve
through all the data points by aid of a flexible ruler and read the
HEUR (or
TSEUR) value off the smooth curve drawn at the interval of
xUR = 0.01. Then the third
derivative quantity
HEUR-UR ( ≡
N (∂HEUR/∂nUR) =
(1−xUR) (∂HEUR/∂xUR)) and the entropy analogue
TSEUR-UR were evaluated and plotted in Fig. 2. The last
quantity signifies, as the above defining equation indicates, the effect of
incoming UR on the actual enthalpic (or entropic) situation of UR in the
solution. Hence,
HEUR-UR (or
TSEUR-UR) provides information about UR - UR interaction
in terms of enthalpy (or entropy). As is evident, there is a clear break
in slope for both
HEUR-UR and
TSEUR-UR at about 0.09. We suggest using the findings from our
1P-probing methodology that urea molecules at this mole fraction start to
aggregate to dimers or trimers, which still form hydrogen bonds directly to the
existing network of H2O. Hence the hydrogen bond
connectivity is retained even above
xUR = 0.01 that is the common
threshold value for other monomer hydrophiles. At
xUR ≈ 0.22, however,
the hydrogen bond network breaks down and Mixing Scheme II sets in, since there
are too many impurity centers to retain the network connectivity. The system
now prepares for “phase separation” at
xUR = 0.266,
the saturation point.
(古賀 精方,稲葉 章)
発 表
古賀 精方,宮崎 裕司,長野 八久,稲葉 章,第44回熱測定討論会(つくば),3A1340 (2008).
Y. Koga, Y. Miyazaki, Y. Nagano, A. Inaba,
J. Phys. Chem. B 112, 11341 (2008).
Copyright © Research Center for Structural Thermodynamics,
Graduate School of Science, Osaka University. All rights reserved.
{kind=link}
{kind=link}