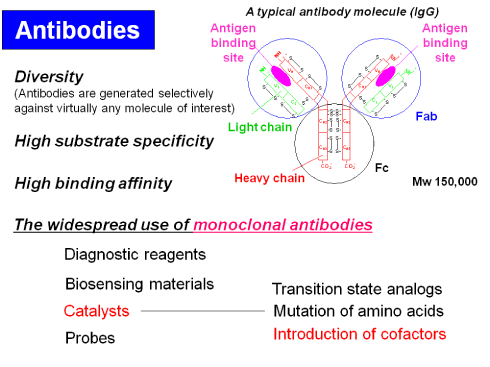
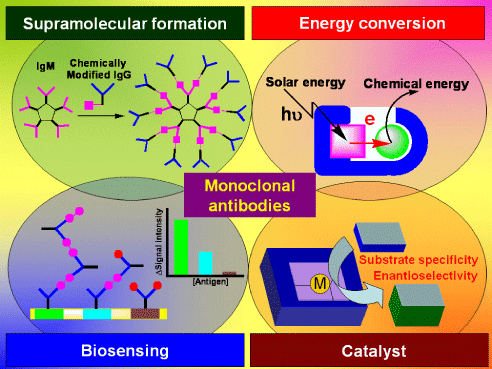
Recent Review of Antibody Chemistry: Let's See Functionalized Antibodies as Biosensing Materials and Catalysts (Highlight Review, 2008)
"Antibody Dendrimer"
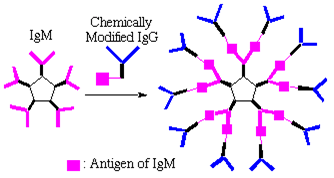
In a complete departure from traditional approaches, Harada used immunoglobin M (IgM) as a core and chemically modified IgGs as branches in a divergent growth strategy which resulted in novel, highly branched antibody based supramolecules with molecular weights of over 2 000 000. Even though these structures were held together by non-covalent bonds, their dendritic nature permitted the binding of antigens with high specificity. These antibody-based dendrimers show great promise for high sensitivity detection of a wide range of chemicals and biomolecules
Reported by C. J. Hawker et al. in J. Mater. Chem., 2003, 13, 2653-2660.
Direct Observation of DNA Catenanes by Atomic Force Microscopy; The first example of the direct observation of DNA catenanes
Yamaguchi, H.; Kubota, K.; Harada, A. Chem. Lett. 2000, 29, 384–385.

DNA catenane Cyclic DNA monomer
The topological structures of DNA catenanes have been studied by AFM and electrophoresis. The single molecular images of DNA catenanes are clearly observed by AFM using a tapping mode at room temperature and in an ambient atmosphere. The topological structures of naked DNA catenanes are directly and successfully observed by AFM without any coating. Our experiment is the first example of the direct observation of DNA catenanes.
Stellate Macroscopic Crystals from Cationic and Anionic Porphyrins
Yamaguchi, H.; Harada, A. Chem. Lett. 2001, 30, 778–779.

Mixing tetrasodium [meso-tetrakis(4-sulfonatophenyl)porphinato]zinc (Zn-TSPP) and meso-tetrakis(4-(trimethylammonio)phenyl)porphine tetratosylate (TTMAPP) in dimethylsulfoxide leads to formation of a star-like (stellate) microcrystal. When meso-tetrakis (N-methylpyridinium-4-yl) porphine p-toluenesul-fonate (TMPyP) was used instead of TTMAPP, such a structure was not obtained. The mesoscale supramolecular structure can be controlled by changing the combination of ionic porphyrins.
Correlation between function and structure of antibodies against porphyrins
Yamaguchi, H.; Kamachi, M.; Harada, A. Angew. Chem. Int. Ed., 2000, 39, 3829-3831.
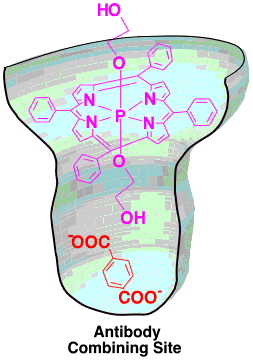
A series of phosphorus(V) porphyrins with axial ligands were prepared. One of the antibodies, 74D7A, recognizes not only the porphyrin (electron donor) but also an acceptor molecule (terephthalic acid) with high selectivity. This antibody binds both porphyrin and an electon acceptor simultaneously in a combining site (as shown in the picture) and thereby facilitates the electron transfer from the porphyrin to the acceptor.
Hydrogen Evolution System Using Antibody-Porphyrin-Complex as Photosensitizer
(1) Onji, T.; Ohara, H.; Yamaguchi, H.; Ikeda, N.; Harada, A. Chem. Lett. 2006, 35, 1126–1127.
(2) Yamaguchi, H.; Onji, T.; Ohara, H.; Ikeda, N.; Harada, A. Bull. Chem. Soc. Jpn. 2009, 82, 1341–1346. BCSJ Award Article
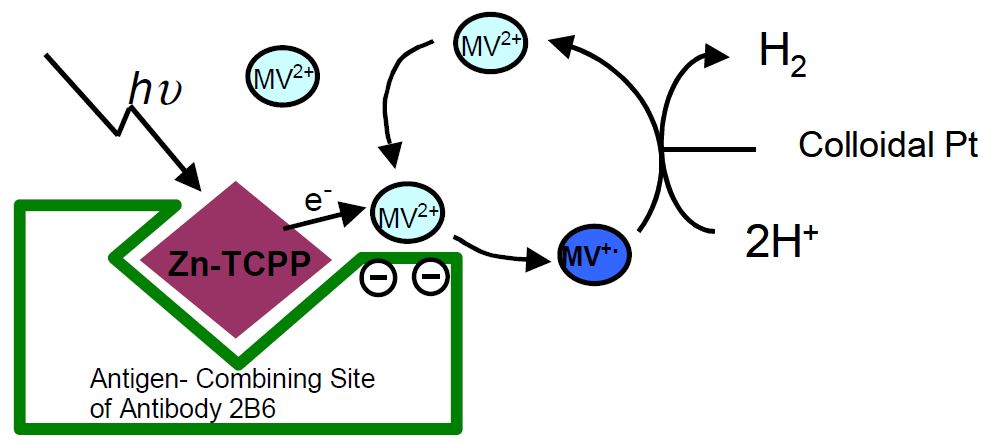
One of the complexes between monoclonal antibody 2B6 for porphyrin and zinc porphyrin (Zn-porphyrin) was used to construct an energy conversion system. The antibody 2B6 could bind Zn-porphyrin with a dissociation constant of 2.1 x 10--8 M, estimated by ELISA. When antibody 2B6 was added to an aqueous solution of Zn-porphyrin, the lifetime of its singlet exited state was lengthened from 1.7 to 2.1 ns. The triplet state lifetime of Zn-porphyrin was also lengthened from 0.5 to 1.2 ms.
Continuous light irradiation of Zn-porphyrin was performed in the presence of the antibody and MV. The color of the solution turned blue and a product with a maximum absorbance at 602 nm appeared. Methyl viologen cationic radical was obtained by the photoinduced electron transfer from porphyrin in the binding site of the antibody to MV. The half-life of methyl viologen cationic radical was over 15 min. No color changes were observed without antibodies. The antibody was found to catalyze the electron transfer to give a stable cationic radical. We found that the cationic radical obtained in the antibody–porphyrin complex system could be utilized for producing chemical energy, hydrogen. The antibody–porphyrin complex, MV, and colloidal Pt were equipped for the construction of a hydrogen evolution system. An increase of the concentration of hydrogen was observed in the solution of the antibody–porphyrin complex. The electron transfer could take place efficiently from porphyrin in the antibody binding site to MV outside of the binding pocket.
Linear Antibody Supramolecules: Application for a Novel Biosensing Method
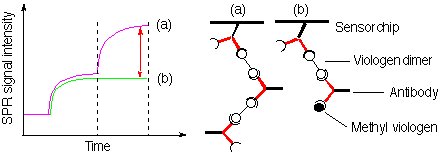
In this study, methyl viologen (MV) is selected as one of the target molecules to be detected. Although viologens are harmful, they are well-known functional molecules which act as herbicides and electron acceptors. MV has been suggested as a potential etiologic factor in Parkinson’s disease because of the structural similarity to the known dopaminergic neurotoxicant, 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine. Anti-viologen antibodies may be expected to be useful not only as a highly sensitive reagent for viologens but also as a functional material to control the electron transfer from electron donors to viologens. However, the detection of viologens at low concentrations is difficult owing to the low sensitivity (small response) in a common SPR biosensor technique using corresponding antibodies. To improve the sensitivity, it is important to detect MV as a large response signal caused by the antibody bindings. A solution to this problem is thought to be the inhibition of the supramolecular formation between the anti-viologen antibody and viologen dimer by MV. A small amount of MV can be detected as a decrease in signal enhancement due to the inhibition of the complex formation between viologen dimer and antibody by MV, compared with the signal intensity of complete supramolecular formation between the antibody and viologen dimer. The sensitivity in this system was 140-fold larger than that in the simple addition of MV to the antibody immobilized to the surface of the sensor chip.
Preparation of monoclonal antibodies capable of selectively detecting explosive substances
Matsumoto, T.; Yamaguchi, H.; Kamijo, K.; Akiyoshi, M.; Matsunaga, T.; Harada, A. Bull. Chem. Soc. Jpn. 2013, 86, 198–202.
BCSJ Award Article
A monoclonal antibody (mAb) specific to triacetone triperoxide (TATP) was produced from the stable hybridoma cell line 15B-1E, generated by the fusion of myeloma cells with spleen cells isolated from a mouse immunized with a conjugate of 3,3′-(9,12-dimethyl-7,8,10,11,13,14-hexaoxaspiro[5.8]tetradecane-9,12-diyl)dipropanoic acid (DHTDA) with the carrier protein, keyhole limpet hemocyanin. The mAb, 15B-1E, bound DHTDA with a dissociation constant (KD) of 4.7 × 10−7 M. The binding of the mAb to TATP was examined by a competitive indirect enzyme-linked immunosorbent assay (ciELISA) and a surface plasmon resonance (SPR) assay. The KD value of mAb 15B–1E to TATP was 5.2 × 10−5 M. Using an mAb raised against DHTDA, we found that 1.8 µM TATP could be detected using mAb 15B–1E.
Substrate selective peroxidation by monoclonal antibody-porphyrin complex
We reported preparation of monoclonal antibodies (mAbs 03-1) against (meso-tetrakis(4-carboxyphenyl)porphyrin, TCPP) and found that these antibodies bind TCPP strongly. The mAbs 03-1 binds not only TCPP but also TCPP-M (Cu, Zn, Fe).1-3 In contrast to the oxidations of various substrates by Horseradish peroxidase (HRP), the complexes of antibody 03-1 with TCPP-M were found to catalyze oxidation of pyrogallol selectively.4。
(1) Harada, A.; Okamoto, K.; Kamachi, M.; Honda, T.; Miwatani, T. Chem. Lett. 1990, 19, 917–918.
(2) Harada, A.; Okamoto, K.; Kamachi, M. Chem. Lett. 1991, 20, 953–956.
(3) Harada, A.; Shiotsuki, K.; Fukushima, H.; Yamaguchi, H.; Kamachi, M. Inorg. Chem. 1995, 34, 1070–1076.
(4) Harada, A.; Fukushima, H.; Shiotsuki, K.; Yamaguchi, H.; Oka, F.; Kamachi, M. Inorg. Chem. 1997, 36, 6099–6102.
Peroxidase Activity of Metalloporphyrin-Antibody Complex
Yamaguchi, H.; Tsubouchi, K.; Kawaguchi, K.; Horita, E.; Harada, A. Chem. Eur. J. 2004, 10(23), 6179-6186.
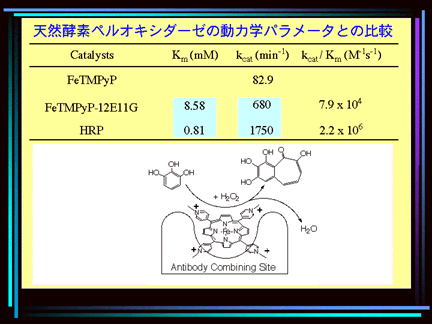
An interesting development is the use of natural macromolecules as carriers for artificial catalysts. One example is the cationic meso-tetrakis(4-N-methylpyridyl)porphyrin and its analogues, which are known to bind to DNA. H. Yamaguchi and A. Harada (Osaka, Japan) bound the FeII complex of this porphyrin to the monoclonal antibody 12E11G. This macromolecular metal complex displays remarkable activity in the oxidation of catechol, guajacol, and pyrogallol in the presence of H2O2, and exhibits a peroxidase activity that lies in the same order as that of horseradish peroxidase while showing a better stability than the natural system.
Reported by Dieter Wohrle in Angew. Chem. Int. Ed., 2005, 44, 7500-7502.
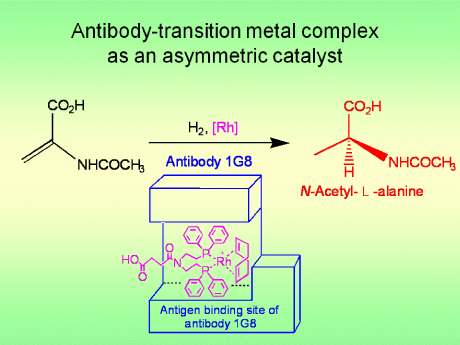
The First Example of the Enanitioselective Hydrogenation by Immunoglobulin-Transition Metal Complex with Substrate Specificity
Yamaguchi, H.; Hirano, T.; Kiminami, H.; Taura, D.; Harada, A. Org. Biomol. Chem. 2006, 4(19), 3571-3573.
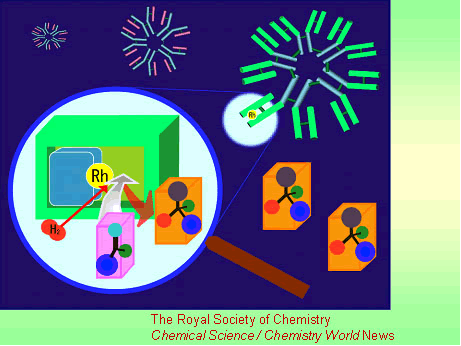
A highly selective catalyst that works like an artificial enzyme has been made using the molecule-targeting system that nature uses to combat infection.
The catalyst, an antibody-rhodium complex, was made by Akira Harada and colleagues at Osaka University, Japan. Harada says his new catalyst successfully combines the reactivity of rhodium with enzyme-like levels of selectivity. Although metals like rhodium, platinum and palladium form some of the most highly active catalysts used by chemists, enzymes with these metals in their active site are not found in nature. Artificial enzymes based on these metals could revolutionise catalysis, said Harada.
The researchers made the catalyst by treating immune system cells with a simple rhodium compound. This process triggers the cells to produce proteins called antibodies, which grab the compound as if it were an invading virus or bacteria. The antibody encloses the rhodium to form an asymmetric environment like an enzyme active site, which controls how substrates approach the metal to give the catalyst its selectivity.
Harada used the antibody-rhodium complex to try to reduce a carbon-carbon double bond in a series of amino acids precursors. The complex proved to be highly selective - only one of the precursors reacted, and only one of two possible enantiomers (mirror-images) of the amino acid product was formed.
The work was welcomed by Neil Thomas, an expert in catalytic antibodies at the University of Nottingham, UK. 'Harada's work provides one of the best examples of combining an organometallic catalyst with a bespoke protein cavity to create an asymmetric catalyst,' he said.
The next challenge is to design a binding site for substrates for which it is difficult to obtain enantiopure products using traditional chiral transition metal catalysts, said Harada.
Reported by Prof. Dr. James Mitchell Crow, in Chemical Science (Royal Society of Chemistry)
Direct Chiral Separation of Binaphthyl Derivatives Using Atroposelective Antibodies
Adachi, T.; Odaka, T.; Harada, A.; Yamaguchi, H. ChemistrySelect 2017, 2, 2622-2625.
Cover Picture

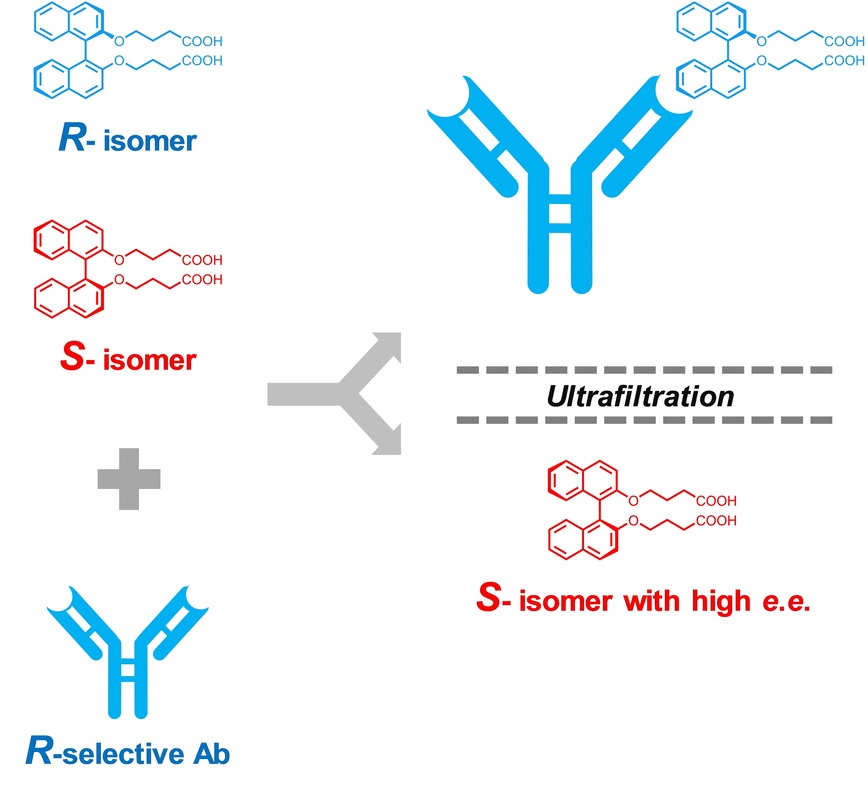
An operationally-simple and rapid methodology for chiral separation was developed. The solutions of the mixture of BN (rac) and atroposelective antibodies were subjected to ultrafiltration to afford purified BN. The difference of molecular weight of mAbs and BN enables the direct use of intact antibodies without any pre-processing or immobilization of mAbs on chromatographic supports. In addition, both enantiomers of BN can be obtained by this procedure where mAbs are prepared for each enantiomer.
Visualization of Chiral Binaphthyl Recognition by Atroposelective Antibodies with Thermoresponsive Polymers
Odaka, T.; Adachi, T.; Harada, A.; Yamaguchi, H. Chem. Lett. 2017, 46, 1173-1175.
Editor's Choice
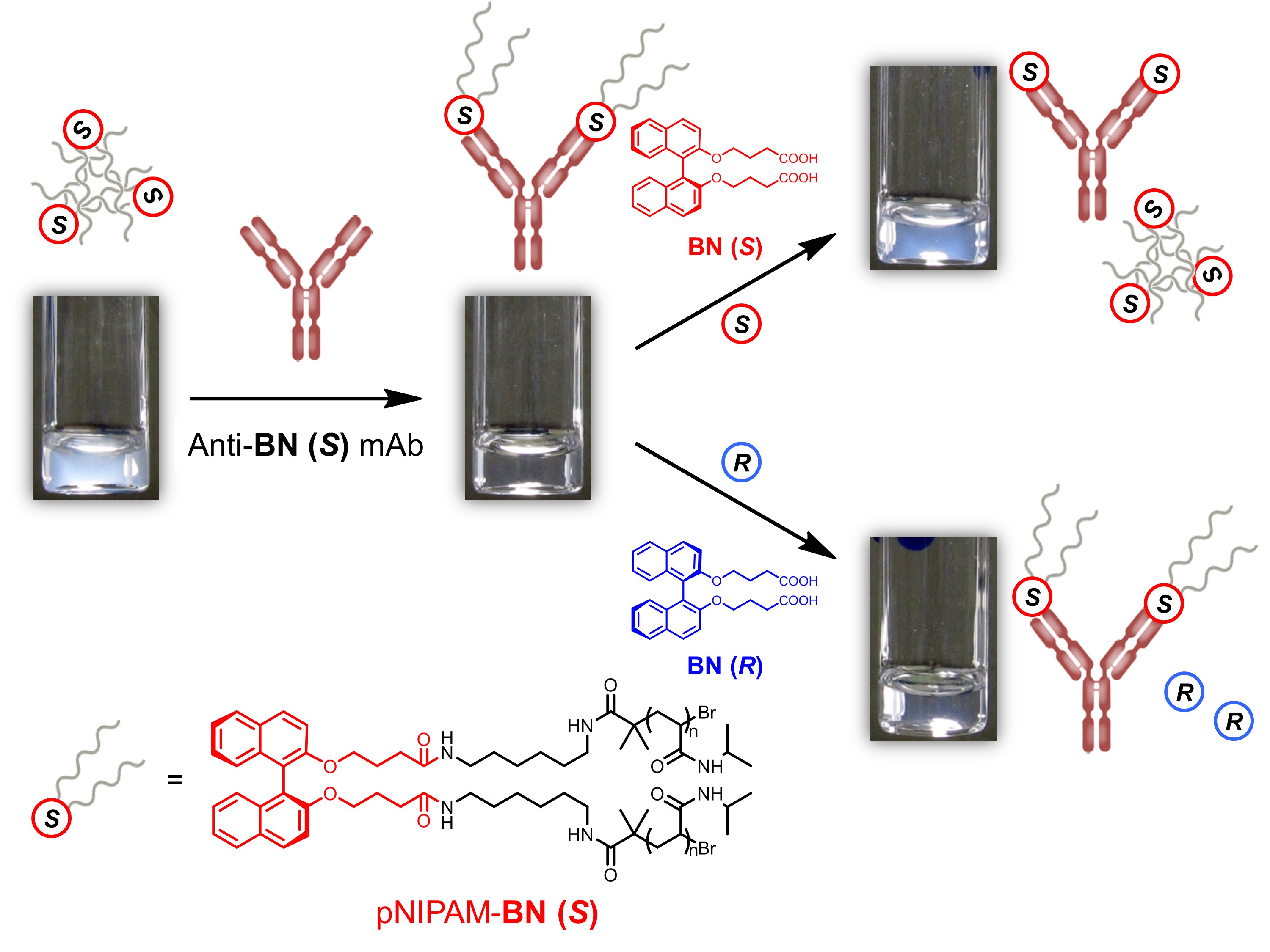
Chiral recognition ability of atroposelective antibodies is applied in chiral sensing. The mAb S1E11 even recognizes the chirality of BN attached on a thermoresponsive polymer, poly-N-isopropylacrylamide (pNIPAM). The aqueous solution of pNIPAM bearing BN (S) (pNIPAM-BN (S)) is cloudy at 32 °C. However, the addition of mAb S1E11 resolves the turbidity by increasing the lower critical solution temperature from 31.5 °C to 33.0 °C. The binding of mAb S1E11 alters the hydration behavior of pNIPAM-based polymer. The addition of BN (S) to the mixture of pNIPAM-BN (S) and mAb S1E11 reclouds the solution, whereas BN (R) does not cause any change. It is suggested that mAb S1E11 switches the binding partner from pNIPAM BN (S) to BN (S). Modification of pNIPAM to BN (S) allows visualization of association and dissociation of mAb S1E11 to pNIPAM-BN (S) by the naked eye. Furthermore, the presence of chiral BN is atroposelectively detected just adding BN to the mixture of pNIPAM-BN (S) and mAb S1E11. Rapid and operationally simple chiral detection system is developed.
Photoswitchable gel assembly based on molecular recognition
Yamaguchi, H.; Kobayashi, Y.; Kobayashi, R.; Takashima, Y.; Hashidzume, A.; Harada, A. Nat. Commun. 2012, 3, 603.

The formation of effective and precise linkages in bottom-up or top-down processes is important for the development of self-assembled materials. Self-assembly through molecular recognition events is a powerful tool for producing functionalized materials. Photoresponsive molecular recognition systems can permit the creation of photoregulated self-assembled macroscopic objects. Here we demonstrate that macroscopic gel assembly can be highly regulated through photoisomerization of an azobenzene moiety that interacts differently with two host molecules. A photoregulated gel assembly system is developed using polyacrylamide-based hydrogels functionalized with azobenzene (guest) or cyclodextrin (host) moieties. Reversible adhesion and dissociation of the host gel from the guest gel may be controlled by photoirradiation. The differential affinities of α-cyclodextrin or β-cyclodextrin for the trans-azobenzene and cis-azobenzene are employed in the construction of a photoswitchable gel assembly system.
Reversible self-assembly of gels through metal-ligand interactions
Kobayashi, Y.; Takashima, Y.; Hashidzume, A.; Yamaguchi, H.; Harada, A. Sci. Rep. 2013, 3, 1243.
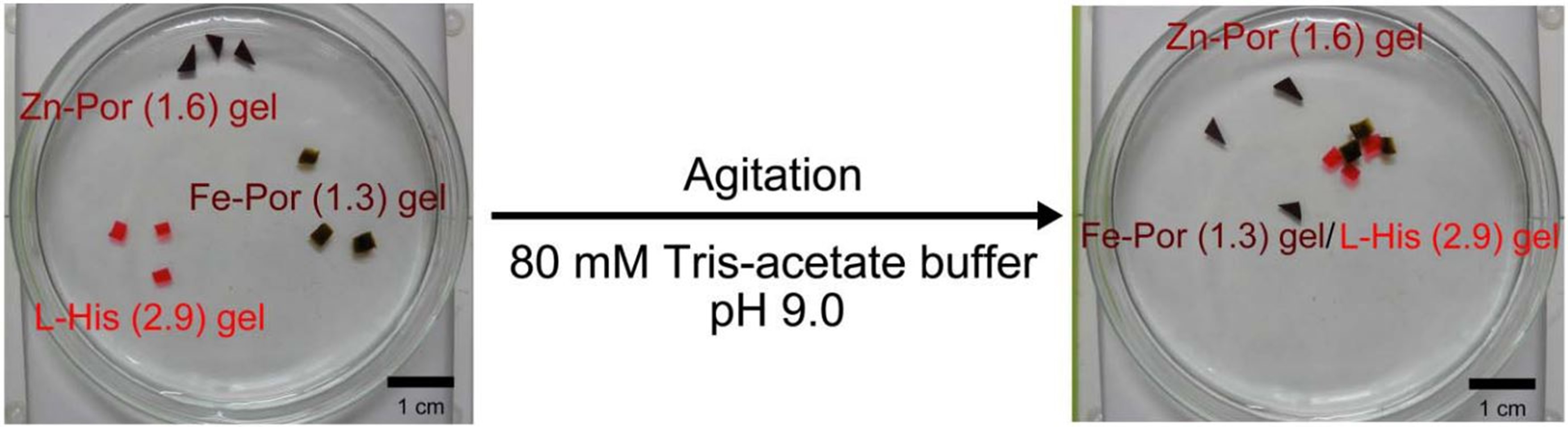
Metal-ligand interactions with various proteins form in vivo metal assemblies. In recent years, metallosupramolecular approaches have been utilized to forge an assortment of fascinating two- and three-dimensional nano-architectures, and macroscopic materials, such as metal-ligand coordination polymeric materials, have promise in artificial systems. However to the best of our knowledge, the self-assembly of macroscopic materials through metal-ligand interactions has yet to be reported. Herein we demonstrate a gel assembly formed via metal-ligand interactions using polyacrylamide modified with Fe-porphyrin and L-histidine moieties. The stress values for the assembly increase as the concentration of Fe-porphyrin or L-histidine in the gels increases. Moreover, agitation of Fe-porphyrin gel, Zn-porphyrin gel, and L-histidine gel in an 80 mM Tris-acetate buffer (pH 9.0) results in selective adhesion of the Fe-porphyrin gel to the L-histidine gel based on the affinities of Fe-porphyrin and Zn-porphyrin with L-histidine.
Manual control of catalytic reactions: Reactions by an apoenzyme gel and a cofactor gel
Kobayashi, Y.; Takashima, Y.; Hashidzume, A.; Yamaguchi, H.; Harada, A. Sci. Rep. 2015, 5, 16254.
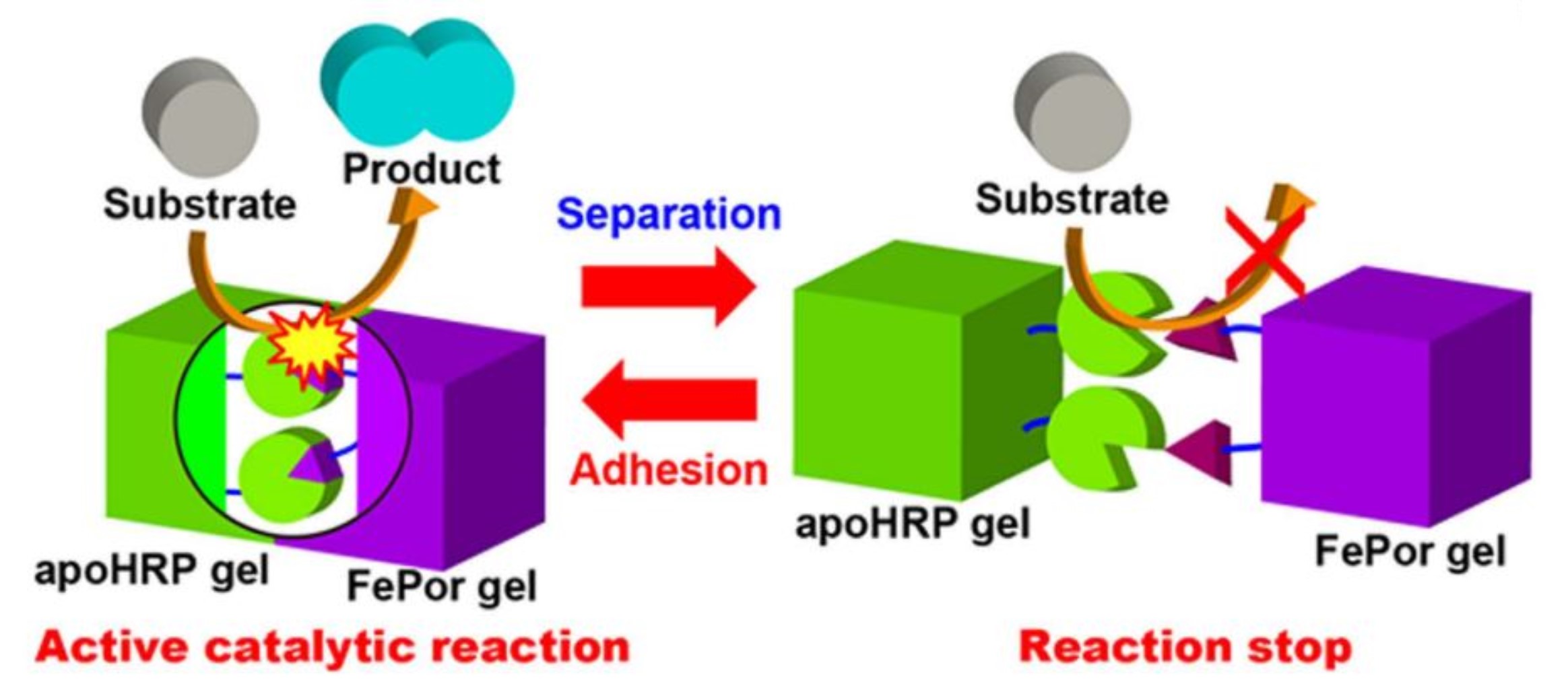
Enzymes play a vital role in catalysing almost all chemical reactions that occur in biological systems. Some enzymes must form complexes with non-protein molecules called cofactors to express catalytic activities. Although the control of catalytic reactions via apoenzyme–cofactor complexes has attracted significant attention, the reports have been limited to the microscale. Here, we report a system to express catalytic activity by adhesion of an apoenzyme gel and a cofactor gel. The apoenzyme and cofactor gels act as catalysts when they form a gel assembly, but they lose catalytic ability upon manual dissociation. We successfully construct a system with switchable catalytic activity via adhesion and separation of the apoenzyme gel with the cofactor gel. We expect that this methodology can be applied to regulate the functional activities of enzymes that bear cofactors in their active sites, such as the oxygen transport of haemoglobin or myoglobin and the electron transport of cytochromes.
Control of Photoinduced Electron Transfer Using Complex Formation of Water-Soluble Porphyrin and Polyvinylpyrrolidone
Cao, Y.; Takasaki, T.; Yamashita, S.; Mizutani, Y.; Harada, A.; Yamaguchi, H. Polymers, 2022, 14, 1191.
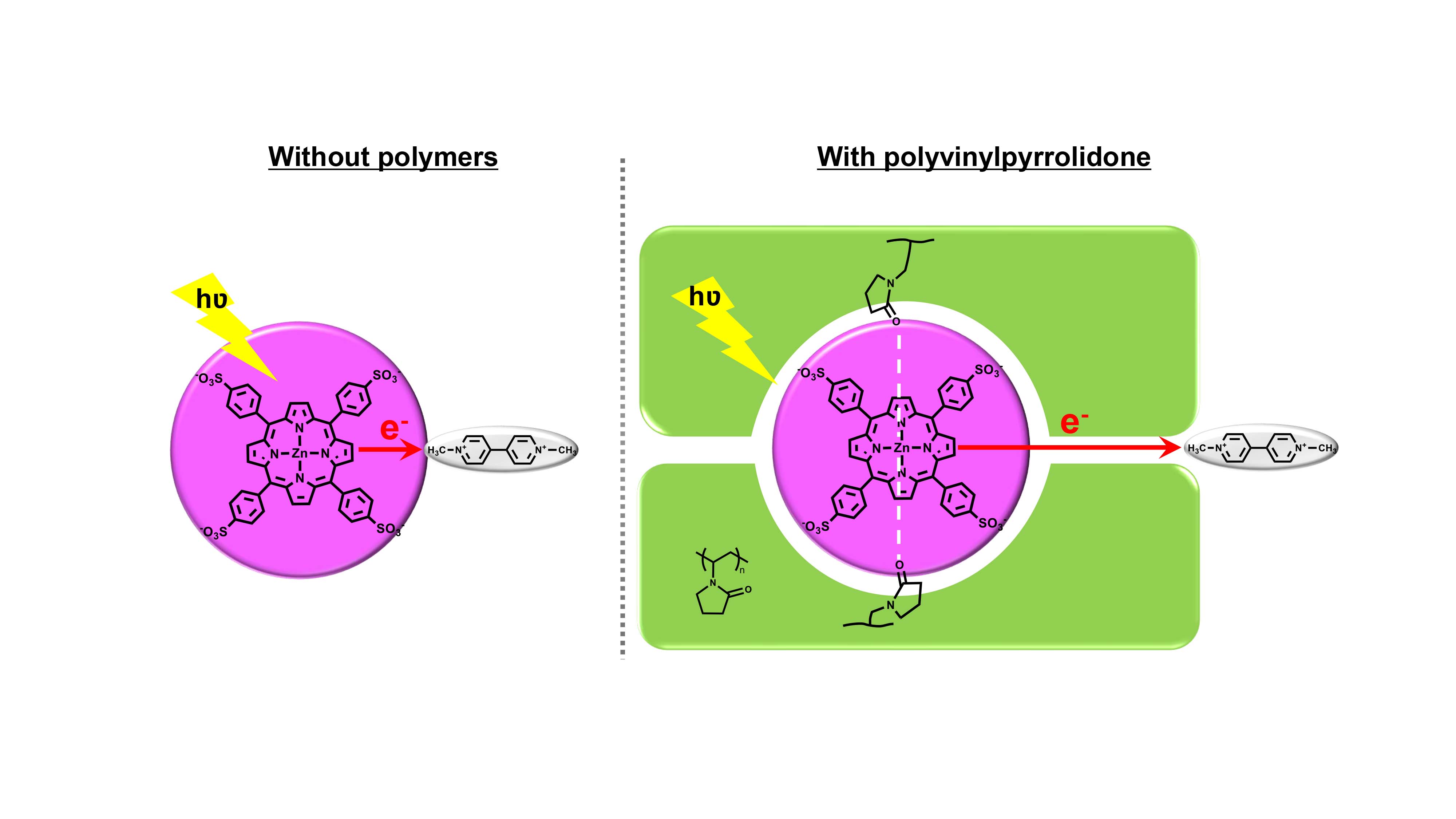
Inspired by the natural photosynthetic system in which proteins control the electron transfer from electron donors to acceptors, in this research, artificial polymers were tried to achieve this control effect. Polyvinylpyrrolidone (PVP) was found to form complex with pigments 5,10,15,20-tetrakis-(4-sulfonatophenyl) porphyrin (TPPS) and its zinc complex (ZnTPPS) quantitatively through different interactions (hydrogen bonds and coordination bonds, respectively). These complex formations hinder the interaction between ground-state TPPS or ZnTPPS and an electron acceptor (methyl viologen, MV2+) and could control the photoinduced electron transfer from TPPS or ZnTPPS to MV2+, giving more electron transfer products methyl viologen cationic radical (MV+•). Other polymers such as PEG did not show similar results, indicating that PVP plays an important role in controlling the photoinduced electron transfer.
Controlled Photoinduced Electron Transfer via Triplet in Polymer Matrix Using Electrostatic Interactions
Cao, Y.; Sotome, H.; Kobayashi, Y.; Ito, S.; Yamaguchi, H. J. Photochem. Photobiol. A, 2024, accepted.
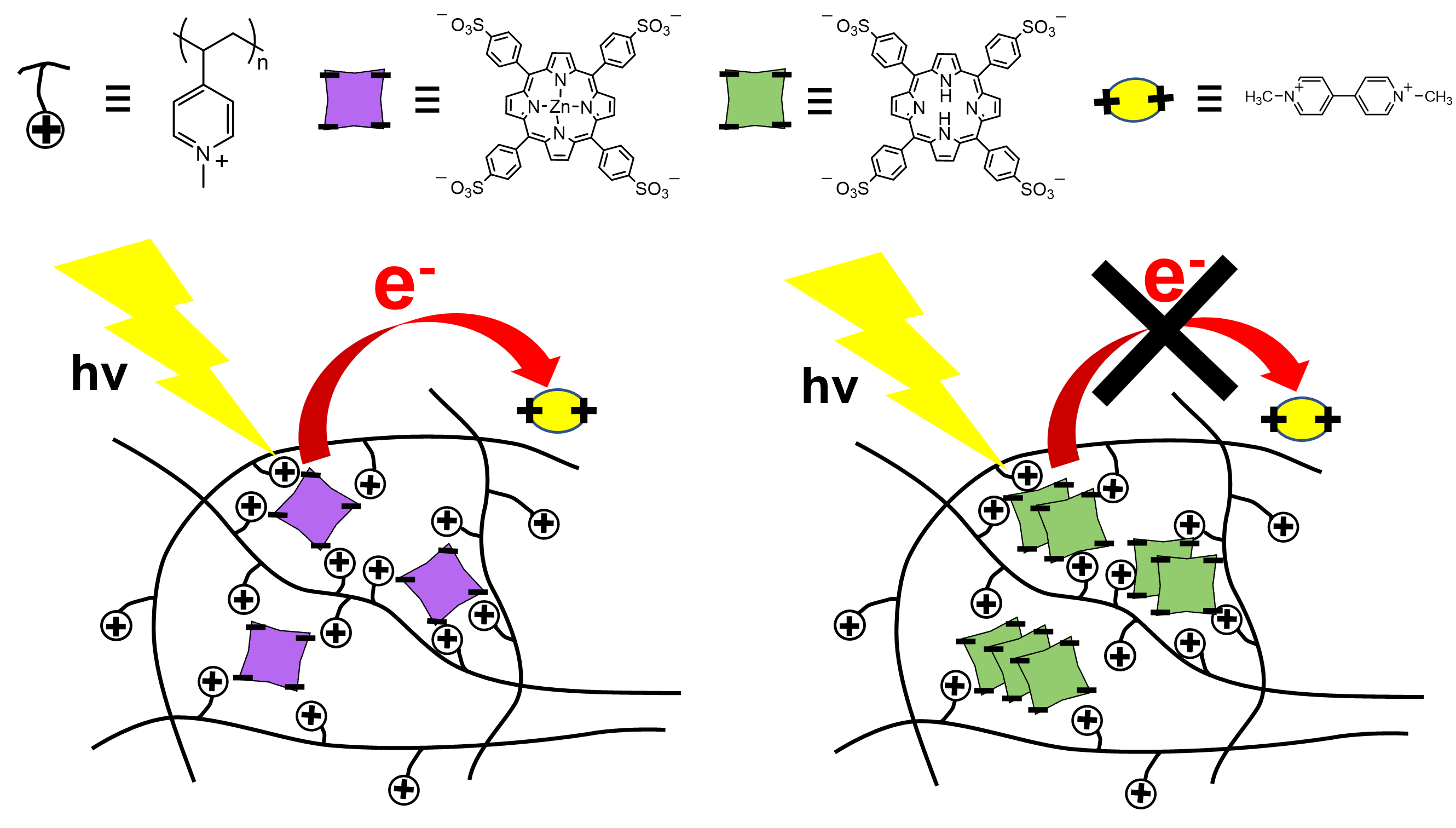
Drawing inspiration from highly efficient natural photosynthetic systems, we developed an artificial photoinduced electron transfer (PET) system within a poly(N-methyl-4-vinylpyridinium) (P4VPMe) polymer matrix. This system features 5,10,15,20-tetrakis-(4-sulfonatophenyl) porphyrin (TPPS) and its zinc complex (ZnTPPS) as electron donors and methylviologen (MV2+) as the electron acceptor. In the presence of an excess of P4VPMe, ZnTPPS exists as individual molecules rather than self-aggregating, favoring PET. Under these conditions, P4VPMe inhibits the formation of ground-state charge-transfer complex between ZnTPPS and MV2+. This shifts the main PET process from singlet to triplet states. ZnTPPS in an excess of P4VPMe demonstrates over a tenfold increase in catalytic activity for the photoinduced oxidation of MV2+ compared to ZnTPPS alone.
Development of Atroposelective Antibodies by Immunization with a Racemic Mixture of Binaphthyl Derivatives
Adachi, T.; Harada, A.; Yamaguchi, H. Bull. Chem. Soc. Jpn. 2019, 92, 1462-1466.
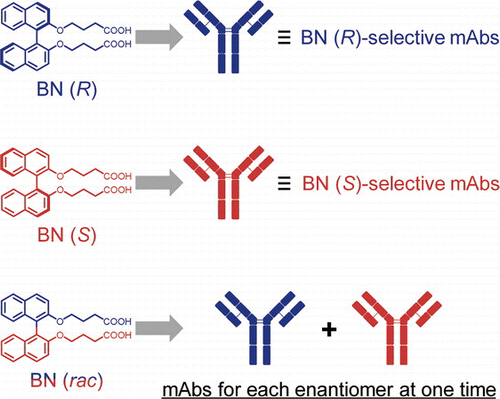
The design and creation of chiral recognition elements are important for the synthesis, separation, and detection of chiral molecules. We prepare monoclonal antibodies (mAbs), which are chemically homogeneous antibodies, as tailored chiral recognition elements by immunizing mice with a racemic mixture of a binaphthyl derivative (BN (rac)) conjugated to keyhole limpet hemocyanin (KLH). Immunization with BN (rac) induces an immunoresponse that is as strong as that with enantiomerically pure antigens and yields mAbs for each enantiomer of BN, simultaneously. Notably, one of the mAbs prepared by immunization with BN (rac) recognizes the axial chirality of the BN enantiomer with a 14000-fold difference in affinity. These findings provide a strategy to obtain atroposelective antibodies for each enantiomer of BN with a single immunization. mAbs also recognize the axial chirality of 1,1’-bi-2-naphthol (BINOL) and 1,1’-binaphthyl-2,2’-diyl hydrogen phosphate (BNPA), which are an important chiral building block and a chiral organic catalyst, respectively. The cross reactivity of mAbs for the partial structure of BN is significantly low. Therefore, mAbs recognize the axial chirality of BN, BINOL, and BNPA by binding their binaphthyl moiety.
Atroposelective antibodies as a designed protein scaffold for artificial metalloenzymes
Adachi, T.; Harada, A.; Yamaguchi, H. Sci. Rep. 2019, 9, 13551.
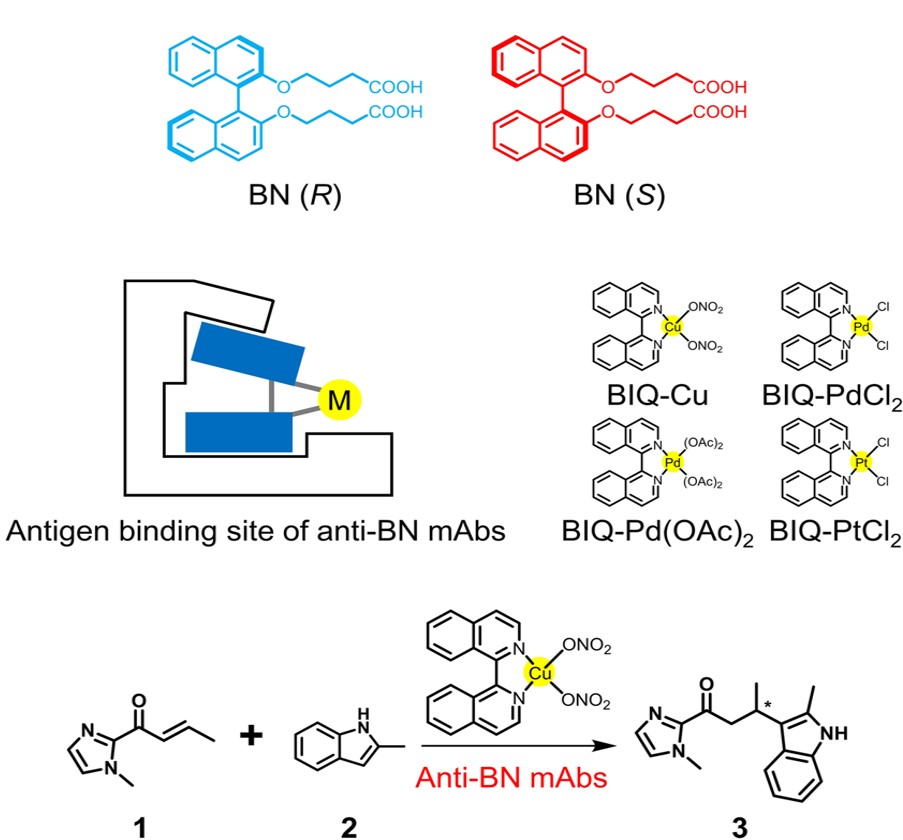
Design and engineering of protein scaffolds are crucial to create artificial metalloenzymes. Herein we report the first example of C-C bond formation catalyzed by artificial metalloenzymes, which consist of monoclonal antibodies (mAbs) and C2 symmetric metal catalysts. Prepared as a tailored protein scaffold for a binaphthyl derivative (BN), mAbs bind metal catalysts bearing a 1,1’-bi-isoquinoline (BIQ) ligand to yield artificial metalloenzymes. These artificial metalloenzymes catalyze the Friedel-Crafts alkylation reaction. In the presence of mAb R44E1, the reaction proceeds with 88% ee. The reaction catalyzed by Cu-catalyst incorporated into the binding site of mAb R44E1 is found to show excellent enantioselectivity with 99% ee. The protein environment also enables the use of BIQ-based catalysts as asymmetric catalysts for the first time.
Mechanical properties of supramolecular polymeric materials cross-linked by donor–acceptor interactions
Itano, M.; Kobayashi, Y.; Takashima, Y.; Harada, A.; Yamaguchi, H. Chem. Commun. 2019, 55, 3809-3812.
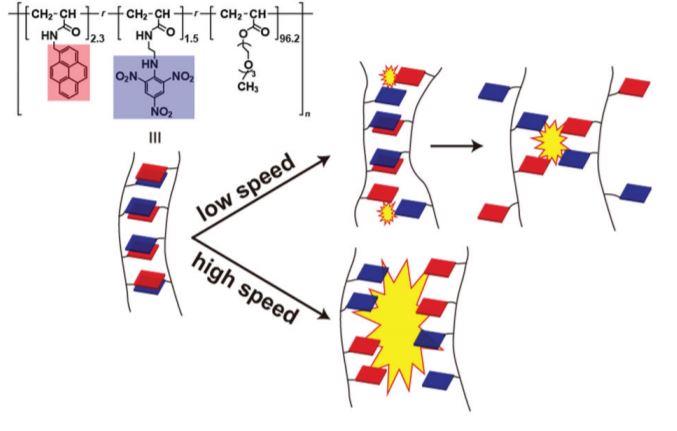
In this paper, we prepare an elastomer with aromatic donor and acceptor molecules introduced on the same polymer side chain. Pyrene (Py), trinitrobenzene (TNB), and poly(methyl triethylene glycol acrylate) (TEG) are employed as a donor unit, acceptor unit, and polymer backbone, respectively. The resultant polymer was dissolved in solution, and then this polymer solution was dried to obtain DA elastomer (Py–TNB TEG elastomer). The fracture energy of the Py–TNB TEG elastomer is higher than that of the covalently cross-linked elastomer (TEG elastomer). Note that the fracture energy of Py–TNB TEG elastomer is further increased by heating and increasing the load speed.
Self-healing and shape-memory properties of polymeric materials cross-linked by hydrogen bonding and metal–ligand interactions
Kobayashi, Y.; Hirase, T.; Takashima, Y.; Harada, A.; Yamaguchi H. Polym. Chem. 2019, 10, 4519-4523.
Back Cover
We prepared supramolecular polymeric materials based on the self-assembly of a tritopic polymer using the combination of hydrogen bonding and metal–ligand interactions. We synthesized poly(δ-valerolactone)–polylactic acid (PVL-PLA) copolymer containing 2-ureido-4(1H)-pyrimidinone (UPy) and 2,2′-bipyridine (bpy) ([M(bpy)n]2+-UPy film) as polymer parts. The fracture energy of the [M(bpy)n]2+-UPy film is higher than that of the polymeric material without non-covalent interactions prepared by bpy-PVL-PLA (bpy film). The fracture energy of the [M(bpy)n]2+-UPy film depends on the type of metal ion added. Note that the [Fe(bpy)3]2+-UPy film showed both self-healing and shape memory properties.
Supramolecular complex formation of polysulfide polymers and cyclodextrins
Kobayashi, Y.; Harada, A.; Yamaguchi H. Chem. Commun., 2020, 56, 13619-13622.
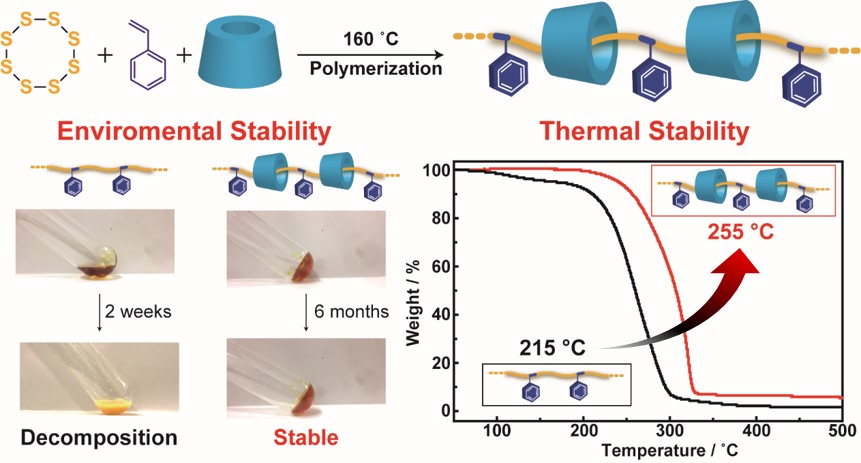
We report the first preparation of a supramolecular polysulfide polymer, which is a polyrotaxane containing sulfur–styrene copolymer and methylated α-cyclodextrins (TMαCDs) as linear and cyclic molecules, respectively (SPRx). Compared to the sulfur–styrene copolymer prepared by a copolymerization method typically used to synthesize polysulfide polymers, the environmental and thermal stabilities of SPRx are significantly improved because the polysulfide polymer is covered with TMαCD.
Supramolecular polysulfide polymers with metal-ligand interactions
Yamagishi, Y.; Kobayashi, Y.; Horiguchi, A.; Kitano, D.; Yamaguchi, H. ChemistrySelect, 2022, 7, e202103991.
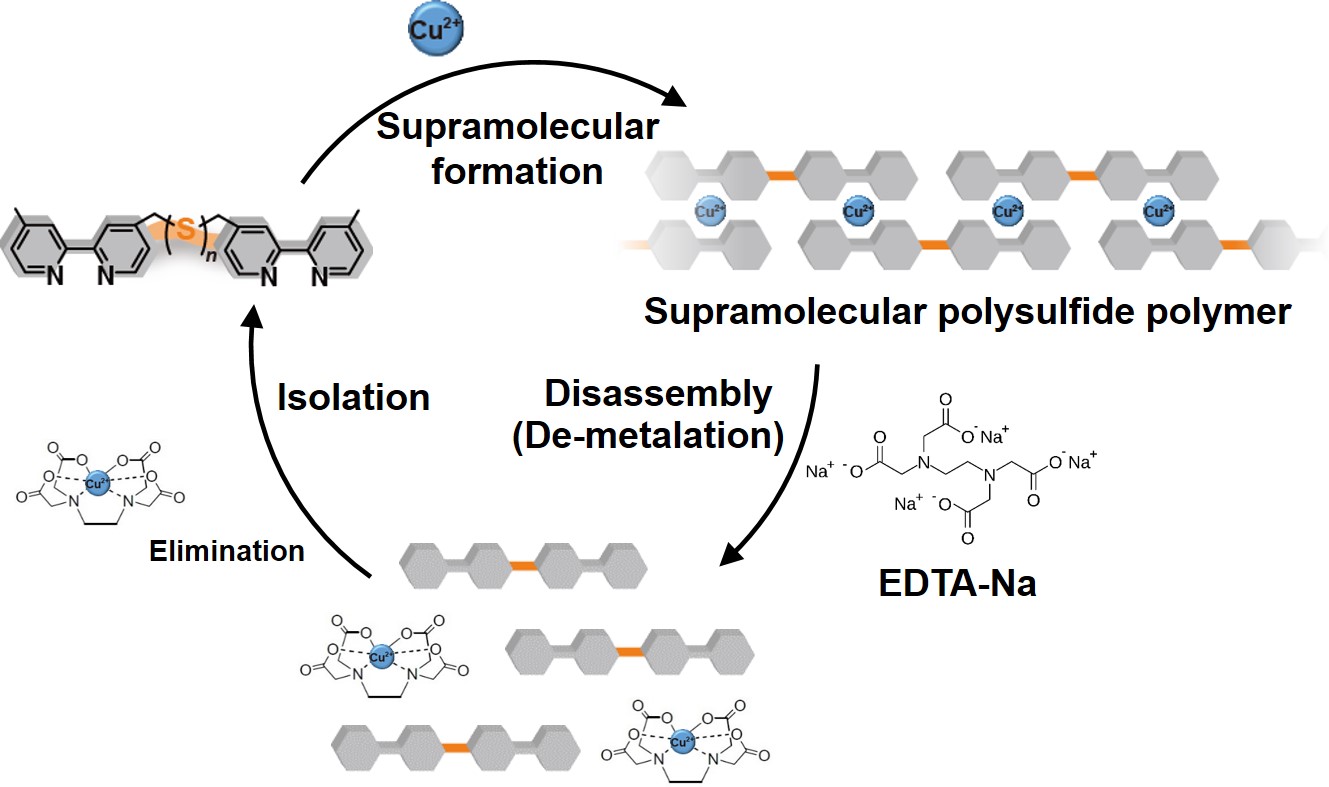
A supramolecular polysulfide polymer using metal-ligand interactions was synthesized for the first time in the world. The supramolecular polysulfide polymer was prepared by introducing 2,2’-bipyridine (bpy) at both ends of linear sulfur and linking the bpy with Cu2+. Disassembly and re-assembly of the supramolecular polysulfide polymers were controlled by de-metalation of Cu2+ from the complex between bpy and Cu2+ and re-formation of the complex with the addition of chelating agents and Cu2+, respectively.
Supramolecular sulfur-containing polymers with hydrogen bonding
Kobayashi, Y.; Yamagishi, Y.; Nishimura, R.; Xiao, C. L.; Kitano, D.; Horiguchi, A.; Hashimoto, S.; Yamaguchi, H. J. Sulfur Chem., 2023, accepted
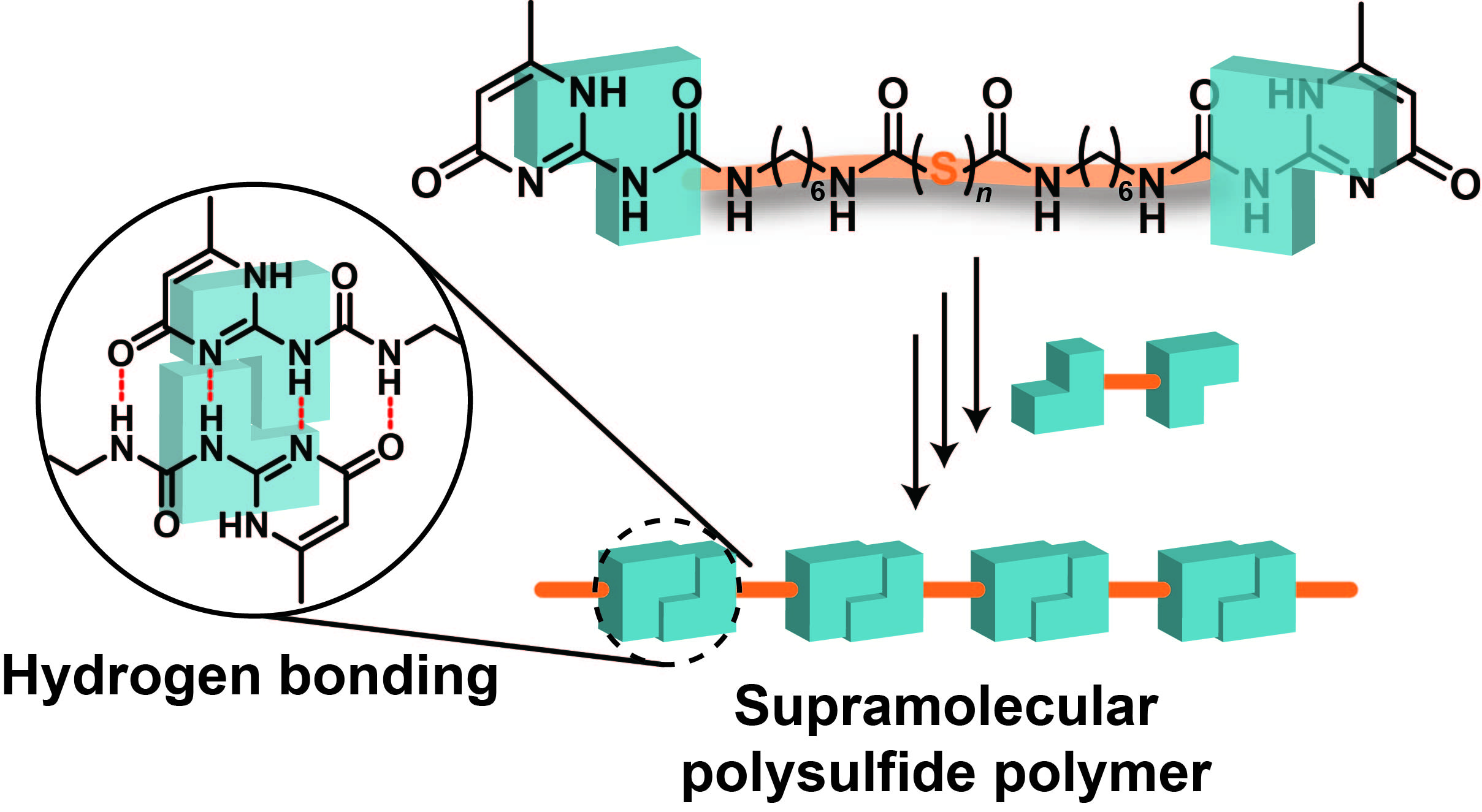
Although sulfur-containing polymers have been realized by various methods such as copolymerization and reverse vulcanization, there are few reports on the synthesis of supramolecular sulfur-containing polymers in which a supramolecular polymer is fused with a sulfur-containing polymer. Herein, we prepare a supramolecular sulfur-containig polymer by introducing a 2-ureido-4[1H]pyrimidinone (UPy) unit at both ends of linear sulfur and connecting between the UPys via hydrogen bonding.
Supramolecular polysulfide polymers cross-linked by metal-ligand interactions
Kobayashi, Y.; Kitano, D.; Nishimura, R.; Yamagishi, Y.;Horiguchi, A.; Yamaguchi, H. Polym. Chem., 2023, accepted
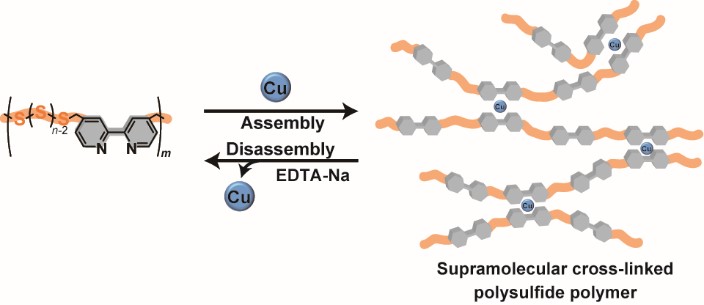
We report the first synthesis of a supramolecular cross-linked polysulfide polymer containing bipyridine (poly(LS-bpy)) using metal-ligand interactions. Complexing the bipyridine part in the poly(LS-bpy) and a metal ion realizes a supramolecular cross-linked polysulfide polymer. The molecular weight of the supramolecular cross-linked polysulfide polymer is 29 times higher than that of a polysulfide polymer obtained from conventional polymerization. In addition, the addition of chelating agents and a metal ion can control the disassembly and assembly of the supramolecular cross-linked polysulfide polymer, respectively. The supramolecular cross-linked polysulfide polymer remarkably improves the thermal stability, which should be useful for applications as a polysulfide polymer material.
Efficient cyclization of linear polymer with pseudopolyrotaxane assistance.
Tsuji, Y.; Kobayashi, Y.; Chunlin, X.; Harada, A.; Yamaguchi, H. Chem. Lett., 2023, 52, 1-4.
Xiao, C.-L.; Kobayashi, Y.; Tsuji, Y.;Harada, A.; Yamaguchi, H. ACS Macro Lett., 2023, accepted.
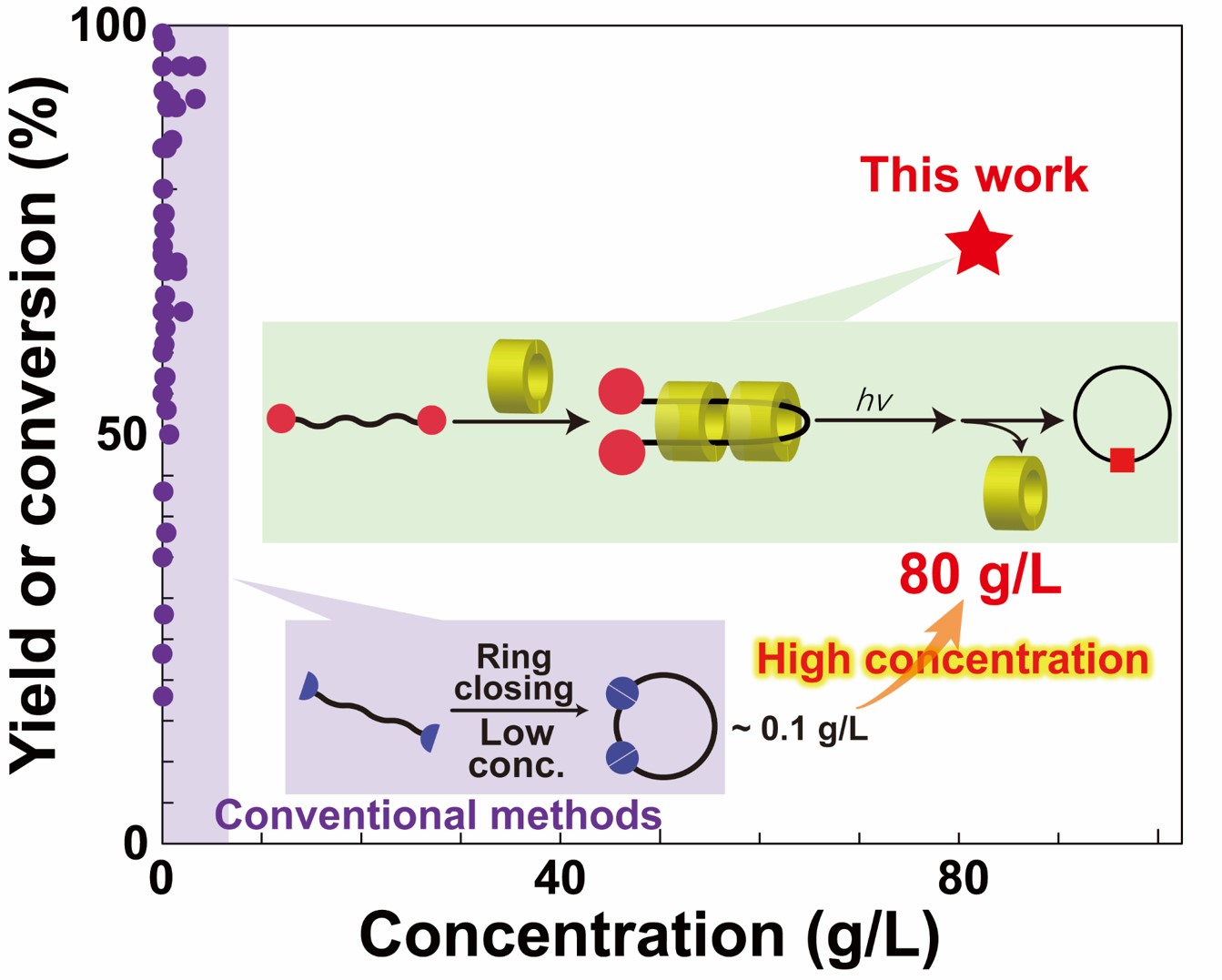
The physical properties of cyclic polymers differ from those of linear polymers. To prepare cyclic polymers, the ends of a linear polymer must intramolecularly react. Cyclic polymers are typically synthesized at low concentration because intermolecular linkage is preferential to intramolecular linkage at high concentrations. Therefore, preparation of cyclic polymers at high concentrations is difficult. This study demonstrates a new approach to obtain cyclic polymers from highly concentrated polymer solutions using pseudo-polyrotaxanes (pPRxs). Cyclic polymers are prepared by penetrating a linear polymer into the cavity of a cyclic host to form pPRx and then the ends of the linear polymer are subsequently connected. Our approach allows the synthesis of cyclic polymers at concentration that do not allow conventional cyclic polymer synthesis. Specifically, although without pPRx, almost no cyclic polymers were obtained (<1%), with pPRx, the cyclic polymer was obtained as the main product (proportion of cyclic PEG 14%) (Chem. Lett.). Afterwards, by appropriately selecting the end groups and cyclic molecules, the yield increased to 70% (ACS Macro Lett.).